A recent case report published in JAAD Case Reports highlights a rare manifestation of porphyria cutanea tarda (PCT) stemming from undiagnosed hereditary hemochromatosis (HH) caused by a HFE gene mutation known as H63D. The study details the case of a 21-year-old man in the United States who developed PCT due to this genetic mutation, shedding light on a lesser-reported aspect of the disease.
Hereditary hemochromatosis is characterized by excessive iron absorption and accumulation, particularly in the liver, and is recognized as a predisposing factor for PCT. The condition arises from mutations in the HFE gene, which encodes a protein essential for regulating iron absorption. While the C282Y mutation is more commonly associated with HH, the H63D mutation has been identified in fewer PCT cases.
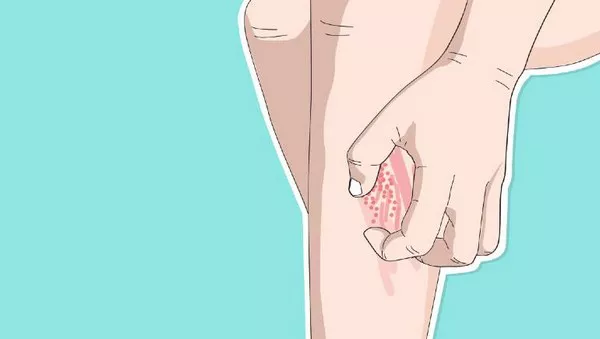
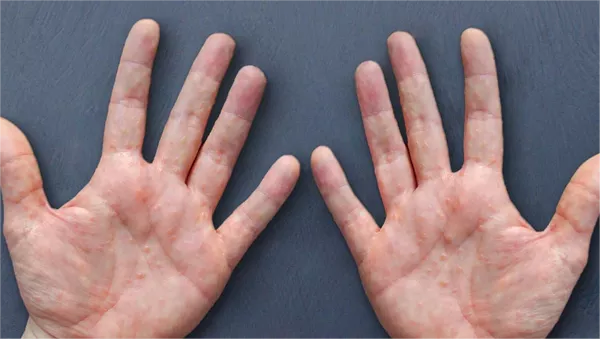
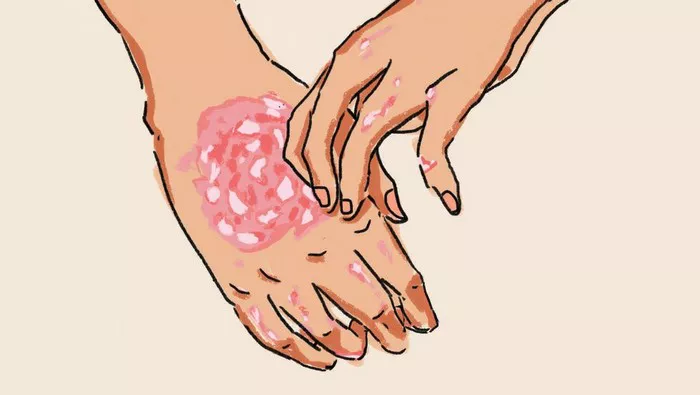
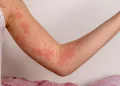
PCT itself results from deficient activity of the uroporphyrinogen decarboxylase enzyme in the liver, leading to the accumulation of porphyrins, which are photosensitive molecules. This accumulation manifests as skin sensitivity to sunlight and the formation of blisters upon exposure.
The patient presented with recurrent skin blisters on his hands and fingers over a three-month period, prompting further investigation. Laboratory tests, including genetic analysis, confirmed homozygosity for the H63D mutation in the HFE gene, thus establishing the diagnosis of hereditary hemochromatosis as the underlying cause of his PCT. Elevated ferritin levels and liver enzyme markers corroborated the iron overload characteristic of HH.
Treatment involved rigorous phlebotomies to reduce iron levels in the blood, aiming to alleviate symptoms and prevent disease progression. Subsequent monitoring showed normalization of ferritin and hemoglobin levels, leading to complete remission of the skin manifestations associated with PCT.
This case underscores the critical role of iron metabolism in the pathogenesis of PCT and highlights the efficacy of therapeutic phlebotomy in managing associated symptoms. The findings contribute valuable insights into the interplay between genetic predisposition and environmental triggers in rare hematologic disorders.
The researchers emphasize the importance of early recognition and management of hereditary hemochromatosis, particularly in individuals presenting with unusual dermatologic symptoms such as those observed in this case. Future studies may further elucidate the complex mechanisms linking iron metabolism and porphyria, potentially guiding more targeted therapeutic approaches.
In conclusion, this case report not only expands our understanding of the genetic basis of PCT but also underscores the significance of comprehensive genetic testing and tailored treatment strategies in managing complex hematologic conditions.
Related Topics: